Noticias General
Inmunoterapia específica con alérgenos con enfermedades autoinmunes concomitantes
La inmunoterapia con alérgenos (ITA) es la única intervención capaz de modificar la historia natural de las enfermedades alérgicas. Sin embargo, su uso en pacientes con enfermedades autoinmunes sigue generando dudas clínicas relevantes.
Clásicamente, la relación entre alergia y autoinmunidad se ha interpretado bajo el paradigma Th1/Th2: mientras que las enfermedades alérgicas se asocian a una respuesta inmunitaria predominantemente Th2, las enfermedades autoinmunes se han vinculado a un predominio Th1. Desde esta perspectiva, cabría esperar una relación inversa entre ambas entidades. No obstante, la evidencia acumulada no ha sido consistente, y este modelo se considera actualmente una simplificación excesiva de la compleja red de mecanismos inmunopatológicos implicados.
La preocupación sobre la posible inducción o exacerbación de enfermedades autoinmunes durante el tratamiento con ITA ha surgido a partir de la descripción de casos aislados, incluyendo Síndrome de Sjögren, esclerosis múltiple, esclerodermia localizada, pericarditis recurrente o vasculitis reportados durante el curso de la ITA1.
En este contexto, revisamos críticamente la evidencia disponible para orientar una práctica clínica basada en evidencia y más personalizada.
Un estudio observacional basado en registros nacionales (1997-2006) en la población danesa, estudió a 18.841 sujetos que recibieron inmunoterapia subcutánea (ITSC) y los comparó con 428.848 sujetos que recibieron tratamiento médico antialérgico convencional. El objetivo principal fue investigar la asociación de ITSC con la incidencia de enfermedades autoinmunes y cardiopatía isquémica. El resultado obtenido de esta observación durante 10 años demostró que la incidencia de enfermedades autoinmunes en la población con ITSC fue menor que la observada en la población con tratamiento convencional2.
Otro estudio realizado en Polonia, en el cual analizaron a 1.011 pacientes que recibieron inmunoterapia sublingual (ITSL) durante 3 años, y les siguieron durante 5 años más para evaluar la incidencia de enfermedades autoinmunes. Los resultados fueron comparados con 1.237 sujetos que no recibieron ITSL y no tenían alergia respiratoria. Al finalizar el período de observación, la población que recibió ITSL presentó una incidencia más baja de enfermedades autoinmunes en comparación con la población control3.
Con respecto al agravamiento de la enfermedad autoinmune durante el curso de la ITA, la evidencia clínica disponible es limitada. Un estudio de serie de casos de sujetos con enfermedades reumatológicas que recibieron ITSL para el tratamiento de la rinitis alérgica, observó que ITSL podía administrarse sin empeorar la actividad inflamatoria de la enfermedad. Algunos pacientes presentaron eventos adversos mayoritariamente leves y sin relación causal demostrada con la ITA4.
La EAACI, en su posicionamiento sobre contraindicaciones clínicas de la ITA5,6, señala que los pacientes con enfermedades autoinmunes deben evaluarse de manera individualizada, valorando fundamentalmente la actividad de la enfermedad, la estabilidad clínica y el balance riesgo/beneficio, ya que la evidencia disponible es limitada y se basa en gran medida en casos aislados y series pequeñas. En estudios observacionales de registros nacionales, la incidencia de autoinmunidad no fue superior en los pacientes tratados con ITA frente a controles, e incluso fue menor en algunas cohortes2,3. Sin embargo, en pacientes con enfermedad autoinmune preexistente la evidencia sigue siendo insuficiente para establecer recomendaciones universales, por lo que la decisión debe ser personalizada y, cuando proceda, coordinada con el especialista de referencia.
En conjunto, la autoinmunidad no debe considerarse una contraindicación para la ITA, sino una situación que requiere valoración clínica individualizada. La decisión terapéutica debe basarse en la actividad y estabilidad de la enfermedad autoinmune y el balance entre riesgos y beneficios para cada paciente.
Bibliografía
- Linneberg A, Madsen F, Skaaby T. Allergen-specific immunotherapy and risk of autoimmune disease. Curr Opin Allergy Clin Immunol. 2012 Dec;12(6):635-9. doi: 10.1097/ACI.0b013e3283588c8d. PMID: 22914311.
- Linneberg A, Jacobsen RK, Jespersen L, Abildstrøm SZ. Association of subcutaneous allergen-specific immunotherapy with incidence of autoimmune disease, ischemic heart disease, and mortality. J Allergy Clin Immunol. 2012 Feb;129(2):413-9. doi: 10.1016/j.jaci.2011.09.007. PMID: 22004944.
- Bozek A, Mućka S, Miodonska M, Zlik A, Mroz-Dybowska M. Effect of sublingual immunotherapy on clinical and laboratory autoimmunity. 2024 Mar;16(4):235-241. doi: 10.2217/imt-2023-0231. PMID: 38214133.
- Fujioka K, Kasahara A, Kida T, Fujii W, Seno T, Wada M, Kohno M, Kawahito Y. Effectiveness and safety of allergen immunotherapy in patients with allergic rhinitis complicated by rheumatic autoimmune diseases: a case series study. Allergy Asthma Clin Immunol. 2022 Jul 11;18(1):63. doi: 10.1186/s13223-022-00703-0. PMID: 35818067
- Pitsios C, et al. Clinical contraindications to allergen immunotherapy: an EAACI position paper. 2015;70(8):897-909. doi:10.1111/all.12638.
- Guidelines on allergen immunotherapy: Prevention of allergy. Pediatr Allergy Immunol. 2017;28(8):728-745.
Actualización en el tratamiento de la granulomatosis eosinofílica con poliangeítis (GEPA)
La granulomatosis eosinofílica con poliangeítis (GEPA), previamente conocida como síndrome de Churg-Strauss, es una vasculitis sistémica que se caracteriza por eosinofilia periférica y afectación multiorgánica. En la actualidad, es una enfermedad en la que coexisten mecanismos autoinmunes e inflamación tipo 2 mediada por eosinófilos. En los últimos años, el manejo de esta enfermedad ha evolucionado desde estrategias clásicas basadas en glucocorticoides e inmunosupresores hacia un enfoque apoyado en terapias biológicas y en la individualización del tratamiento según fenotipo clínico.
- Fenotipos clínicos
En la última década, se han identificado diferentes fenotipos dentro de la GEPA con diferentes implicaciones en su manejo. El fenotipo ANCA positivo suele acompañarse de manifestaciones vasculíticas como glomerulonefritis, neuropatía periférica o púrpura cutánea. En cambio, los pacientes con fenotipo ANCA negativo tienden a un perfil más marcado por la infiltración eosinofílica, presentando asma de difícil control, infiltrados pulmonares, afectación cardiaca y a una mayor intensidad de inflamación sistémica.
Además de esta clasificación, cada vez se habla más de endotipos inmunológicos en los que la inflamación tipo 2, mediada por eosinófilos y citocinas como IL-4, IL-5 e IL-13, representa un eje fisiopatológico clave en muchos pacientes. Esto resulta especialmente relevante para el alergólogo, ya que permite conectar la GEPA con otras enfermedades eosinofílicas como el asma grave o la rinosinusitis crónica con poliposis nasal.
Desde el punto de vista terapéutico, esto obliga a plantear diferentes estrategias, ya que el componente vasculítico responde mejor a inmunosupresión clásica o a la depleción de linfocitos B, mientras que la inflamación eosinofílica constituye una diana clave para terapias biológicas específicas.
En este contexto, resulta interesante revisar las diferentes opciones terapéuticas:
- Tratamientos
2.1 Glucocorticoides
A día de hoy, los glucocorticoides siguen siendo la base del tratamiento, ya que su rápido efecto sobre la inflamación eosinofílica permite un control eficaz en la mayoría de los pacientes, especialmente en fases iniciales. Sin embargo, su uso prolongado se asocia a numerosos efectos adversos, entre los que destacan osteoporosis, diabetes, infecciones, hipertensión arterial y toxicidad cardiovascular. Además, gran porcentaje de pacientes presenta enfermedad dependiente de corticoides o recaídas al reducirlos, lo que ha impulsado el desarrollo de opciones terapéuticas dirigidas a reducir su uso, uno de los principales objetivos terapéuticos actualmente.
2.2 Inmunosupresores clásicos
En pacientes con enfermedad grave o con afectación renal, cardiaca o neurológica, los inmunosupresores clásicos siguen desempeñando un papel fundamental. En la fase de inducción, la ciclofosfamida se sigue considerando el tratamiento estándar, especialmente en el fenotipo vasculítico, mientras que en la fase de mantenimiento, se utilizan habitualmente azatioprina o metotrexato. Sin embargo, la toxicidad asociada a estos fármacos y la disponibilidad de nuevas alternativas, han relegado su uso a un segundo plano, especialmente en pacientes con predominio eosinofílico.
2.3 Terapias biológicas
2.3.1 Terapias dirigidas contra la vía de la interleucina 5 (IL-5) y su receptor
La IL-5 es fundamental en la diferenciación, activación y supervivencia de los eosinófilos, por lo que es una diana terapéutica clave en la GEPA.
- Mepolizumab
Ha sido el primer anticuerpo monoclonal en demostrar su eficacia, con evidencia clínica tanto en inducción como en mantenimiento y remisión. El ensayo pivotal MIRRA supone un punto de inflexión al demostrar que este fármaco aumenta el tiempo de remisión, reduce las recaídas y disminuye la necesidad de glucocorticoides. Además, estos resultados se han confirmado en estudios observacionales europeos, reforzando su papel como tratamiento de referencia en pacientes con fenotipo eosinofílico.
- Benralizumab
Ha emergido como una de las principales novedades en el tratamiento de la GEPA. Su mecanismo de acción, dirigido contra el receptor de la IL-5, permite una depleción casi completa de eosinófilos. El ensayo clínico MANDARA demuestra que benralizumab es no inferior a mepolizumab en la inducción de la remisión, con un control de la enfermedad y una reducción del uso de glucocorticoides similares. Además, estudios en vida real confirman su eficacia en el control del asma, la reducción de exacerbaciones y la mejoría global de la enfermedad. Por todo ello, se perfila como una opción de primera línea en pacientes con GEPA de predominio eosinofílico, especialmente en aquellos con enfermedad respiratoria o necesidad de optimizar la retirada de corticoides.
- Reslizumab
Evidencia mucho más limitada en GEPA. No dispone de ensayos clínicos específicos y su uso se basa en series de casos y en la extrapolación de datos procedentes del asma eosinofílica. Su papel es marginal y se reserva a pacientes con mala respuesta o intolerancia a otras terapias anti-IL-5.
2.3.2 Terapias dirigidas contra la vía de la interleucina 4 e interleucina 13 (IL-4/IL-13)
Dupilumab es un anticuerpo monoclonal dirigido frente a la subunidad alfa del receptor de interleucina-4 (IL-4Rα), lo que inhibe la señalización de IL-4 y de IL-13, citocinas clave en la respuesta inmunitaria tipo 2. El bloqueo de estas vías permite actuar sobre la inflamación tipo 2 de forma más amplia. En pacientes con GEPA, los datos disponibles proceden de estudios observacionales y sugieren beneficios en el control del asma y de la rinosinusitis crónica, pero el beneficio sobre el componente vasculítico de la enfermedad es incierto.
- Terapias dirigidas contra linfocitos B: Rituximab
Rituximab se mantiene como una herramienta fundamental en el tratamiento de la GEPA, sobre todo en pacientes con ANCA positivos y predominio de enfermedad vasculítica. Depleciona linfocitos B, lo que lo convierte en alternativa a ciclofosfamida, especialmente en pacientes con enfermedad grave, refractaria o con contraindicación a la misma.
- Estrategias terapéuticas actuales: hacia la medicina de precisión
El enfoque actual del tratamiento de la GEPA se basa en una estrategia individualizada que tiene en cuenta la gravedad de la enfermedad, el fenotipo clínico (ANCA vs eosinofílico), los órganos afectados y la necesidad de minimizar la exposición a glucocorticoides. En términos generales:
- En enfermedad no grave, se priorizan terapias biológicas (especialmente anti-IL-5) asociadas a glucocorticoides.
- En enfermedad grave, se requiere la combinación de glucocorticoides sistémicos con inmunosupresores como ciclofosfamida o rituximab.
- En mantenimiento, se selecciona la terapia en función del perfil del paciente, priorizando estrategias ahorradoras de esteroides.
Este enfoque permite optimizar el control de la enfermedad reduciendo la toxicidad a largo plazo.
- Impacto en la calidad de vida
Un aspecto clave en el manejo de la GEPA es que la remisión de la enfermedad no implica la resolución completa de los síntomas. Muchos pacientes mantienen asma persistente, rinosinusitis crónica o fatiga, lo que impacta significativamente en su calidad de vida. Las terapias biológicas dirigidas contra la inflamación tipo 2 han demostrado beneficios claros en este ámbito, lo que refuerza el papel del alergólogo e inmunólogo en el manejo integral de la enfermedad.
- Perspectivas futuras y nuevas dianas terapéuticas
El futuro del tratamiento de la GEPA se orienta hacia un modelo de medicina de precisión basado en la identificación de biomarcadores predictivos de respuesta, la estratificación de los pacientes según endotipos inmunológicos y la optimización de estrategias terapéuticas que permitan reducir o eliminar la exposición a glucocorticoides. Este enfoque permitirá individualizar el tratamiento y mejorar los resultados clínicos a largo plazo.
El creciente conocimiento de la fisiopatología de esta enfermedad ha impulsado la identificación y el desarrollo de nuevas dianas terapéuticas. Tezepelumab, un anticuerpo monoclonal dirigido frente a la TSLP (thymic stromal lymphopoietin), actúa modulando de forma precoz múltiples vías implicadas en la respuesta tipo 2. En el momento actual, su eficacia está establecida en asma grave y su evidencia en GEPA procede de estudios preliminares, pero su mecanismo de acción lo posiciona como una opción prometedora en pacientes con enfermedad refractaria o con inflamación tipo 2 compleja.
Por otro lado, otras estrategias en investigación incluyen inhibidores de JAK, lo que apoya el desarrollo de terapias que actúen sobre niveles precoces y globales de la inflamación. Estos avances insinúan que las opciones terapéuticas se ampliarán en los próximos años.
- Conclusiones
La GEPA ha evolucionado desde un modelo terapéutico basado en glucocorticoides e inmunosupresores hacia un enfoque de medicina personalizada. La consolidación de terapias dirigidas contra la vía de la interleucina-5 (IL-5), junto con la incorporación de nuevas estrategias terapéuticas, ha transformado de forma significativa el manejo de estos pacientes, permitiendo un mejor control de la enfermedad con una menor carga de toxicidad. En este contexto, el principal reto actual radica en integrar estas opciones en una estrategia individualizada que optimice los resultados clínicos y mejore la calidad de vida.
- Bibliografía
Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med. 2017;376(20):1921–1932. doi:10.1056/NEJMoa1702079.
Bettiol A, Urban ML, Dagna L, Cottin V, Franceschini F, Del Giacco S, et al. Mepolizumab for eosinophilic granulomatosis with polyangiitis: a European multicenter observational study. Arthritis Rheumatol. 2022;74(2):295–306. doi:10.1002/art.41943.
Wechsler ME, Nair P, Terrier B, Walz B, Bourdin A, Jayne DRW, et al. Benralizumab versus mepolizumab for eosinophilic granulomatosis with polyangiitis. N Engl J Med. 2024;390(10):911–921. doi:10.1056/NEJMoa2311155.
Cottu A, Groh M, Desaintjean C, Marchand-Adam S, Guillevin L, Puéchal X, et al. Benralizumab for eosinophilic granulomatosis with polyangiitis. Ann Rheum Dis. 2023;82(12):1580–1586. doi:10.1136/ard-2023-224624.
Bettiol A, Urban ML, Padoan R, Groh M, Lopalco G, Egan A, et al. Benralizumab for eosinophilic granulomatosis with polyangiitis: a retrospective, multicentre, cohort study. Lancet Rheumatol. 2023;5(12):e707–e715. doi:10.1016/S2665-9913(23)00243-6.
Molina B, Padoan R, Urban ML, Novikov P, Caminati M, Taillé C, et al. Dupilumab for relapsing or refractory sinonasal and/or asthma manifestations in eosinophilic granulomatosis with polyangiitis: a European retrospective study. Ann Rheum Dis. 2023;82(12):1587–1593. doi:10.1136/ard-2023-224756.
Mohammad AJ, Hot A, Arndt F, Moosig F, Guerry MJ, Amudala N, et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg–Strauss). Ann Rheum Dis. 2016;75(2):396–401. doi:10.1136/annrheumdis-2014-206095.
Chung SA, Langford CA, Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation guideline for the management of ANCA-associated vasculitis. Arthritis Rheumatol. 2021;73(8):1366–1383.
Hellmich B, Sanchez-Alamo B, Schirmer JH, Berti A, Blockmans D, Cid MC, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis. 2024;83(1):30–47. doi:10.1136/ard-2022-223764.
López Paraja M, Starita Fajardo G, Donate Velasco I, Lucena López D, Iranzo Alcolea MP, Lirola Sánchez FJ, et al. Molecular pathogenesis and targeted therapies in eosinophilic granulomatosis with polyangiitis: an updated review. Int J Mol Sci. 2025;26(22):11141. doi:10.3390/ijms262211141.
Bettiol A, Emmi G. New therapeutic targets in eosinophilic granulomatosis with polyangiitis. Curr Opin Rheumatol. 2023;35(1):15–21. doi:10.1097/BOR.0000000000000892.
Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384(19):1800–1809. doi:10.1056/NEJMoa2034975.
EAACI abre la convocatoria Member-at-Large 2026-2028
Convocatoria EAACI: Member-at-Large 2026-2028
EAACI ha abierto la convocatoria de candidaturas para Member-at-Large (mandato 2026-2028), cargos que formarán parte del Executive Committee que se elegirá en la Asamblea General de junio de 2026.
Los seis Members-at-Large pueden ser propuestos por cualquier miembro de EAACI y por Sociedades Nacionales, y asumirán tareas específicas para apoyar al Board of Officers y las estrategias presidenciales. Las personas seleccionadas deberán asistir a todas las reuniones de trabajo del periodo 2026-2028, incluidas las primeras reuniones presenciales tras el Congreso (lunes 15 – martes 16 de junio de 2026).
- Fecha límite: domingo, 26 de abril de 2026.
- Requisitos clave: residencia y actividad profesional en Europa, membresía EAACI con cuota 2026 pagada y revisión del Bylaw Art. 16 (conflicto de interés/lealtad).
- Más información y solicitud en este enlace
Síndrome Hiper-IgE (HIES): bases genéticas, características clínicas y el nexo entre inmunodeficiencia y atopia
Los Síndromes de Hiper-IgE (HIES), comprenden un grupo heterogéneo de errores innatos de la inmunidad. Se definen por una tríada clínica característica que incluye niveles séricos de IgE elevados (frecuentemente >2000 UI/mL), eccema crónico de inicio neonatal e infecciones recurrentes, principalmente estafilocócicas y fúngicas en piel y pulmones. Aunque inicialmente se describió como una entidad única, los avances en genética han identificado al menos once defectos genéticos en diez genes distintos que convergen en este fenotipo, aunque con matices clínicos y biológicos distintivos.
El origen del HIES reside en mutaciones genéticas específicas que determinan si la enfermedad se presenta de forma autosómica dominante o recesiva.
1. Clasificación Genética y Mecanismos inmunológicos
1.1 HIES Autosómico Dominante (AD-HIES)
Deficiencia de STAT3: Es la forma más común (prevalencia aproximada de 1 en 1 millón) y está causada principalmente por mutaciones en el gen STAT3. La proteína STAT3 es el nodo central de señalización para múltiples citocinas (IL-6, IL-10, IL-21 e IL-22). La señalización defectuosa de STAT3 impide la activación del factor de transcripción retinoid-related orphan receptor gamma-t (RORγt), esencial para el linaje Th17 (subtipo de linfocitos T CD4+), importantes en la defensa frente a hongos y bacterias extracelulares.
La ausencia de células Th17 conlleva un déficit de producción de IL-17 e IL-22. La IL-17 es imperativa para la quimiotaxis y el reclutamiento de neutrófilos, lo que explica la formación de abscesos «fríos» (ausencia de signos inflamatorios locales) por S. aureus y neumonías recurrentes con formación de neumatoceles. Las infecciones fúngicas, incluyendo candidiasis mucocutánea y aspergilosis pulmonar, también son frecuentes. Mientras que, la deficiencia de IL-22 debilita la integridad de las uniones estrechas epiteliales y reduce la síntesis de péptidos antimicrobianos, facilitando la colonización patogénica y la penetración de alérgenos.
Las vías de señalización de citocinas que involucran Il-6, IL-10 e IL-21 también están gravemente afectadas. El papel crucial de las citocinas IL-6 e IL-21, radica en su capacidad para promover la diferenciación de los linfocitos Th17 a través de la activación de STAT3, el cual induce la transcripción del factor RORγt. La alteración de la señalización de la IL-10, compromete la función de las células T reguladoras (Treg) y rompe el equilibrio homeostático del sistema inmunitario. En consecuencia, se produce una extrema susceptibilidad a infecciones recurrentes y un estado de inflamación crónica y desregulación inmunitaria que caracteriza a la enfermedad.
Las células Th17 además contrarrestan la inflamación alérgica impulsada por IgE. Al estar bloqueada la vía Th17, se produce un sesgo masivo hacia una respuesta Th2, con una sobreproducción de IL-4, IL-5 e IL-13 que estimula el cambio de isotipo a IgE y la hipereosinofilia. Además, STAT3 es mediador de la señalización de la IL-21, citocina que normalmente actúa como un regulador negativo de la síntesis de IgE en los linfocitos B. La interrupción de esta vía de control, sumada a una señalización defectuosa de la IL-10, perpetúa un estado de inflamación alérgica crónica y una producción descontrolada de anticuerpos IgE. El predominio de citocinas Th2 promueve la eosinofilia periférica y la sensibilización de mastocitos, lo que explica la gravedad del eccema y otras manifestaciones alérgicas en estos pacientes.
El eccema suele comenzar durante el período neonatal, antes que la dermatitis atópica común. En la mayoría de los casos, los niveles séricos de IgE están elevados, frecuentemente superiores a 2000 UI/ml. Sin embargo, existen casos con niveles de IgE solo ligeramente elevados, pudiendo incluso situarse dentro del rango normal. La IgE específica contra S. aureus y Candida albicans está elevada, por lo que se considera que la producción de IgE antígeno-específica está aumentada en este trastorno. La eosinofilia está presente en aproximadamente el 90% de los pacientes, y el recuento de eosinófilos en sangre periférica suele ser superior a 700/ml. Los niveles de otras inmunoglobulinas son generalmente normales, y la respuesta de anticuerpos a la vacunación se ve afectada en al menos algunos pacientes con HIES.
A diferencia de otras formas, presenta anomalías esqueléticas y del tejido conectivo.
Deficiencia de IL6ST (GP130) dominante negativo: El gen IL6ST codifica la proteína GP130, una subunidad común para 10 citocinas de la familia IL-6. La IL-6 se considera una de las citocinas proinflamatorias más potentes. Las citocinas de la familia IL-6 tienen funciones pleiotrópicas en la regulación de la reacción de fase aguda, la estimulación de las células B y la regulación del equilibrio entre las células T reguladoras y efectoras. Su deficiencia mimetiza casi todos los síntomas del HIES clásico autosómico dominante. Sin embargo, estos pacientes tienen una respuesta de fase aguda (inflamatoria) retrasada o ausente ante infecciones bacterianas. Presentan escoliosis y osteoporosis, pero a diferencia de STAT3, suelen conservar cierta funcionalidad en la señalización de IL-11, lo que suele evitar la craniosinostosis.
Deficiencia de CARD11 (también conocido como CADINS): CARD11-DN, es esencial para la activación de NF-κB tras la estimulación del receptor de células T (TCR). Mutaciones dominantes negativas en CARD11 interrumpen su señalización. Los defectos sesgan la respuesta hacia el perfil Th2 y defectos en la proliferación de células T y B. Cursan con atopia grave (dermatitis, asma), hipogammaglobulinemia, infecciones virales (molusco, herpes) y respiratorias. Los niveles de IgE son altos, pero generalmente menores que en la deficiencia de STAT3. Tienen mayor riesgo de desarrollar enfermedades autoinmunitarias y tumores malignos.
TGFBR1 y TGFBR2 (Ganancia de Función – GOF): Causan el Síndrome de Loeys-Dietz: Aunque se conoce como un trastorno del tejido conectivo, las mutaciones de ganancia de función (GOF) en los receptores de TGF-β causan un fenotipo de Hiper-IgE. Presentan aneurismas aórticos, tortuosidad arterial, úvula bífida y paladar hendido. La señalización alterada de TGF-β desvía a los linfocitos hacia un perfil Th2, elevando la IgE, causando alergias alimentarias, asma y esofagitis eosinofílica.
Deficiencia de ERBIN (pérdida de función – LOF): ERBIN es una proteína que facilita la comunicación entre STAT3 y la señalización de TGF-β. Su deficiencia causa una actividad excesiva de TGF-β y de la vía IL-4/IL-4R. Solo se han reportado pocos casos. Presentan enfermedades gastrointestinales eosinofílicas graves (EGID), infecciones bacterianas, aneurismas e hipermovilidad articular.
STAT6 (Ganancia de Función – GOF): STAT6 es el principal mediador de la IL-4 y la IL-13, induciendo la diferenciación Th2 y el cambio de clase a IgE en células B. Cursan con enfermedad alérgica grave desde el nacimiento, dermatitis atópica refrctaria, múltiples alergias alimentarias y medicamentosas, y episodios de anafilaxia frecuentes (a veces fatales).
1.2. HIES Autosómico Recesivo (AR-HIES)
Es mucho más raro y suele asociarse a consanguinidad. No presenta las anomalías esqueléticas del AD-HIES, pero tiene mayor incidencia de complicaciones neurológicas y neoplasias. A diferencia de la forma dominante (STAT3), las variantes recesivas suelen clasificarse como Inmunodeficiencias Combinadas, ya que afectan profundamente la supervivencia y función de múltiples linajes de linfocitos.
Deficiencia de DOCK8: Es la causa principal de AR-HIES. DOCK8 es vital para el remodelado del citoesqueleto de actina, la migración celular y la formación de la sinapsis inmunológica. Su ausencia compromete la supervivencia de las células T vírgenes, células NK y la memoria de las células CD8+, limitando la vigilancia antitumoral y la defensa antiviral. Los pacientes presentan un marcado sesgo Th2, lo que deriva en niveles masivos de IgE y alergias alimentarias graves con anafilaxia. Tienen una susceptibilidad extrema a infecciones virales cutáneas (VPH, Herpes, molusco) y un alto riesgo de malignidad (linfomas y carcinomas escamosos).
Deficiencia TYK2: Como parte de la familia JAK, TYK2 media la señalización de IL-6, IL-12, IL-23 e interferones tipo I. Su disfunción afecta la diferenciación Th1 y Th17, lo que explica una susceptibilidad a micobacterias intracelulares (como el BCG), Salmonella e infecciones virales.
Deficiencia PGM3 (Fosfoglucomutasa 3): Afecta la síntesis de precursores de la glicosilación de proteínas (UDP-GlcNAc). La fisiopatología exacta aún se investiga, pero el defecto en la glicosilación de lípidos y proteínas es crítico para la integridad neuronal y el sistema inmune. Se distingue por asociar la deterioro neurológico, ataxia, retraso cognitivo, hipomielinización y displasia esquelética. Cursan con infecciones recurrentes, neutropenia y fallo de médula ósea. A diferencia de DOCK8, suelen mimetizar mucho al tipo STAT3.
IL6ST (Pérdida de Función Parcial o Completa): La deficiencia parcial causa infecciones recurrentes, eccema, craniosinostosis, dientes retenidos. La deficiencia completa resulta en el Síndrome de Stüve-Wiedemann, que es letal y asocia displasia esquelética, disfunción pulmonar neonatal trombocitopenia congénita, dermatitis atópica, anomalías renales y una respuesta de fase aguda defectuosa.
IL6R (Pérdida de Función – LOF): Se manifiesta con dermatitis atópica e infecciones estafilocócicas recurrentes. Un signo característico es la ausencia de respuesta de fase aguda (PCR indetectable) durante infecciones activas
Deficiencia de ZNF341 (Pérdida de Función – LOF): ZNF341 es un factor de transcripción necesario para la producción de la proteína STAT3. Al no haber ZNF341, los niveles de STAT3 son bajos. Su deficiencia mimetiza el AD-HIES STAT3, aunque presenta menor compromiso del tejido conectivo. Presentan los mismos problemas dentales y esqueléticos, aunque suelen tener respuestas inflamatorias más fuertes (fiebre, PCR elevada) y los niveles de células NK son notablemente bajos.
SPINK5 (Síndrome de Netherton): aunque es una enfermedad de la barrera cutánea, se incluye en el espectro HIES por su tríada clínica. Existe un defecto en la descamación de la piel por falta de un inhibidor de proteasas. Se identifica por eritrodermia ictiosiforme, cabello en bambú (trichorrhexis invaginata) y manifestaciones atópicas severas como alergia alimentaria y anafilaxia.
2. Patrones de Sensibilización y Alergia
Aunque comparten niveles masivos de IgE total, los patrones de sensibilización alérgica y la expresión de enfermedades atópicas varían según el defecto genético subyacente.
Los pacientes con deficiencia de STAT3 presentan una paradoja: tienen una IgE total extraordinariamente alta pero una IgE específica baja, lo que resulta en un fenotipo alérgico clínico más leve que en DOCK8. Este fenómeno se atribuye a que la señalización defectuosa de STAT3 afecta primordialmente la diferenciación Th17 y la supresión de IgE mediada por IL-21. Además, la mutación en STAT3 provoca un defecto intrínseco en la degranulación de los mastocitos mediada por el receptor FcεRI. Por tanto, no induce un sesgo Th2 tan extremo como en otras variantes, resultando en un fenotipo alérgico relativamente más moderado.
En la deficiencia de DOCK8, el fenotipo alérgico es mucho más grave y pronunciado, con niveles de IgE específica a alimentos muy elevados y una predisposición mayor a la anafilaxia. Esto ocurre porque la falta de DOCK8 rompe el equilibrio entre las respuestas Th1 y Th2, provocando un exceso de citocinas como IL-4 e IL-13 que fuerzan a las células B a producir IgE ante alérgenos comunes. Además, el reclutamiento masivo de eosinófilos y una barrera cutánea muy dañada facilitan que los alérgenos penetren y sensibilicen al paciente de forma extrema.
Los pacientes con deficiencia de CARD11 y ganancia de función STAT6, también presentan niveles altos de IgE específica contra alimentos y aeroalérgenos que se correlacionan directamente con la clínica de dermatitis atópica, asma y anafilaxia.
En PGM3-LOF se observa dermatitis atópica de inicio temprano y múltiples alergias. Comparten características de STAT3 y DOCK8, aunque la prevalencia de alergia alimentaria es menor que en DOCK8.
En la deficiencia de ERBIN y TGFBR1/2 GOF, existe un aumento de la IgE específica al desviar la diferenciación de células T hacia un perfil Th2. Se asocian fuertemente con enfermedad gastrointestinal eosinofílica (EGID), asma y rinitis.
Los pacientes con Sindrome de Netherton (SPINK5) son propensos a desarrollar manifestaciones atópicas graves, incluyendo alergia alimentaria, rinoconjuntivitis y anafilaxia
3. Manifestaciones No Inmunológicas
Rasgos Multisistémicos: En el AD-HIES STAT3 y en las deficiencias de IL6ST, predominan las anomalías óseas como la fragilidad esquelética con fracturas ante traumas mínimos, la escoliosis progresiva y la hiperextensibilidad articular; sin embargo, la craneosinostosis es un rasgo específico de la deficiencia de IL6ST. Un hallazgo dental casi exclusivo del STAT3-HIES es la retención de dientes de leche por falta de reabsorción radicular, lo que provoca una «doble hilera» dental. En cuanto a la morfología facial, mientras el STAT3-HIES desarrolla con la edad una «facies tosca» (frente prominente y puente nasal ancho), el síndrome de Loeys-Dietz (TGFBR1/2) se distingue por hipertelorismo, úvula bífida o paladar hendido, y la deficiencia de PGM3 suele presentar labios prominentes y fosas nasales anchas. El Síndrome de Netherton (SPINK5) está caracterizado por el «cabello en bambú» e ictiosis. STAT6-GOF, se asocia con una talla baja.
A nivel sistémico, las complicaciones vasculares y neurológicas también varían según el genotipo. El STAT3-HIES muestra una alta predisposición a aneurismas coronarios y arterias cerebrales tortuosas, mientras que en la deficiencia de DOCK8 el riesgo se desplaza hacia eventos agudos como infartos cerebrales, hemorragias y vasculitis. Por último, la deficiencia de PGM3 destaca como la única variante del espectro que asocia un deterioro neurológico intrínseco.
Malignidad y Riesgo Oncológico: El riesgo de cáncer es crítico, especialmente en DOCK8, donde el 17% de los pacientes desarrolla linfomas o carcinomas escamosos (vinculados a virus como EBV y VPH). En STAT3-HIES, predomina el riesgo de linfoma no Hodgkin.
4. Estrategias Diagnósticas
El diagnóstico definitivo del Síndrome de Hiper-IgE (HIES) se fundamenta en la integración de hallazgos clínicos, biomarcadores específicos y la confirmación molecular. El rasgo cardinal es una IgE sérica >2000 UI/mL persistente que, a diferencia de la dermatitis atópica común, no suele fluctuar ante alérgenos en la variante STAT3; junto a esto, un recuento bajo o ausente de células Th17 (típicamente <0.3%) actúa como un marcador patognomónico del defecto en la vía STAT3. Para estandarizar la sospecha, se utiliza el NIH Score, una herramienta que evalúa parámetros como infecciones recurrentes, rasgos faciales y anomalías esqueléticas o dentales, donde una puntuación ≥40 sugiere fuertemente la forma dominante (AD-HIES). Finalmente, la secuenciación genética es el paso esencial para distinguir entre la mutación en STAT3 y las formas recesivas (DOCK8, PGM3, TYK2 o ZNF341), las cuales pueden carecer de rasgos óseos pero presentan un cuadro de inmunodeficiencia combinada grave.
5. Abordaje terapéutico
La profilaxis antimicrobiana, es la piedra angular para prevenir abscesos y daño pulmonar irreversible. Se utilizan de forma crónica trimetoprima-sulfametoxazol e itraconazol /voriconazol. El tratamiento sustitutivo con inmunoglobulinas se inicia si hay defectos en la producción de anticuerpos. Los biológicos como el dupilumab (anti-IL-4R) ha sido transformador para el eccema refractario en STAT3, DOCK8 y ZNF341, mientras que, el omalizumab resulta útil para el control de síntomas alérgicos y asma. El trasplante de Progenitores Hematopoyéticos (TPH), es el tratamiento curativo de elección para DOCK8. En STAT3 es más controvertido y se reserva para complicaciones graves, no corrige los defectos esqueléticos ni del tejido conectivo. Actualmente se encuentra en investigación, la corrección génica mediante CRISPR-Cas9 para STAT3 y DOCK8.
BIBLIOGRAFIA
- Sutanto H, Adytia GJ, Fetarayani D. Hyper IgE Syndrome: Bridging the Gap Between Immunodeficiency, Atopy, and Allergic Diseases. Curr Allergy Asthma Rep. 2025 Mar 14;25(1):17. doi: 10.1007/s11882-025-01196-8. PMID: 40082265.
- AlYafie R, Velayutham D, van Panhuys N and Jithesh PV (2025) The genetics of hyper IgE syndromes. Front. Immunol. 16:1516068. doi: 10.3389/fimmu.2025.1516068
- Minegishi Y. Hyper-IgE syndrome, 2021 update. Allergol Int. 2021 Oct;70(4):407-414. doi: 10.1016/j.alit.2021.07.007. Epub 2021 Aug 18. PMID: 34419355.
- Al-Shaikhly T, Ochs HD. Hyper IgE syndromes: clinical and molecular characteristics. Immunol Cell Biol. 2019 Apr;97(4):368-379. doi: 10.1111/imcb.12209. Epub 2018 Nov 19. PMID: 30264496.
- Boos AC, Hagl B, Schlesinger A, Halm BE, Ballenberger N, Pinarci M, Heinz V, Kreilinger D, Spielberger BD, Schimke-Marques LF, Sawalle-Belohradsky J, Belohradsky BH, Przybilla B, Schaub B, Wollenberg A, Renner ED. Atopic dermatitis, STAT3- and DOCK8-hyper-IgE syndromes differ in IgE-based sensitization pattern. Allergy. 2014 Jul;69(7):943-53. doi: 10.1111/all.12416. PMID: 24898675.
- Schimke LF, Sawalle-Belohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, Rieber N, Cremer R, Maass E, Dopfer R, Reichenbach J, Wahn V, Hoenig M, Jansson AF, Roesen-Wolff A, Schaub B, Seger R, Hill HR, Ochs HD, Torgerson TR, Belohradsky BH, Renner ED. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. J Allergy Clin Immunol. 2010 Sep;126(3):611-7.e1. doi: 10.1016/j.jaci.2010.06.029. Erratum in: J Allergy Clin Immunol. 2010 Nov;126(5):1015. PMID: 20816194.
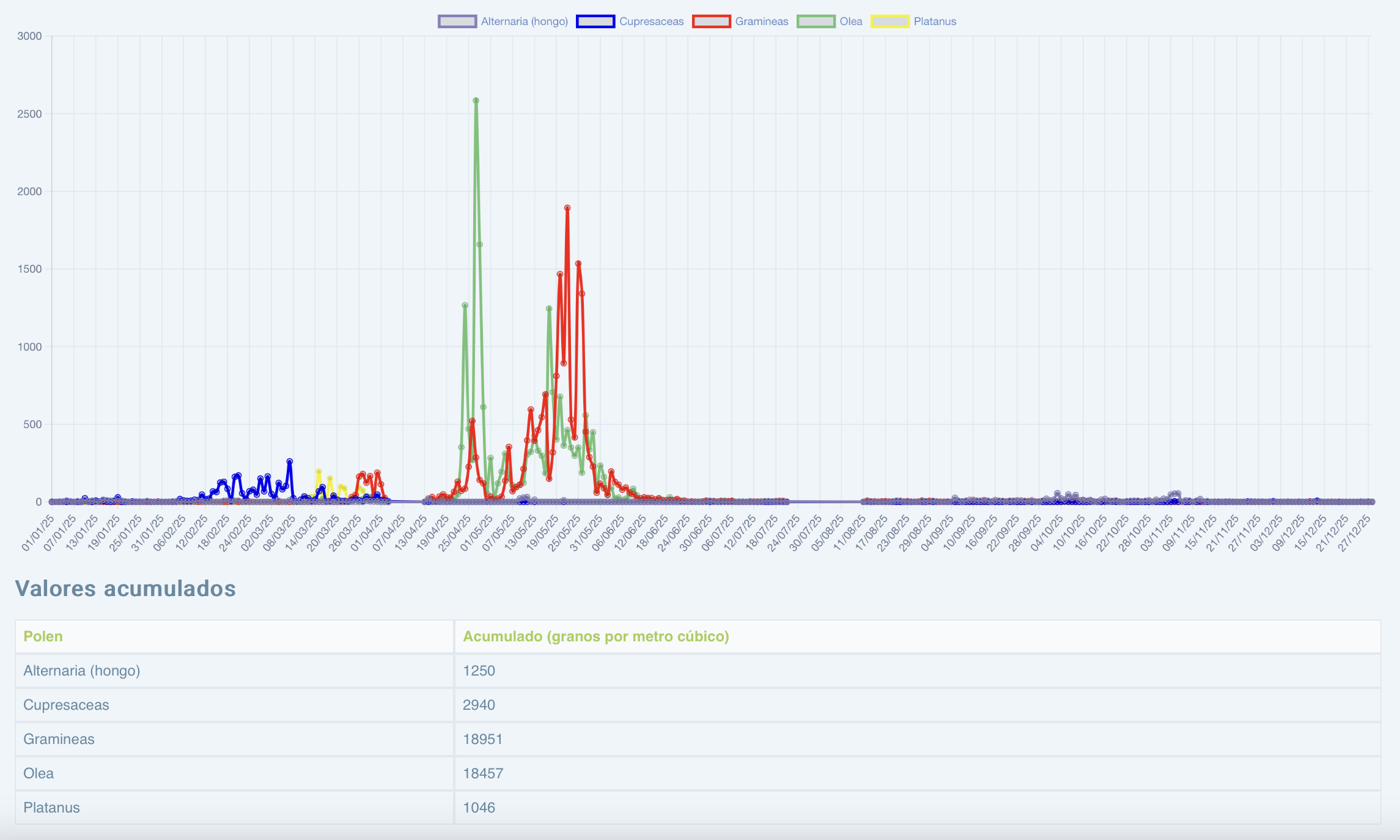
PREVISIONES PÓLENES 2026

El pasado 18 de marzo tuvo lugar la rueda de prensa en la que el Dr. Juan José Zapata, presidente del comité de Aerobiología Clínica, dio a conocer las previsiones de pólenes para esta primavera. Desde este medio ofrecemos un resumen de los datos.
● Las previsiones apuntan a que la primavera, en el conjunto del territorio
español, será intensa por los niveles generalizados de pólenes.
● El “efecto lavado” asociado a las lluvias registradas en los primeros meses
del año reduce temporalmente el polen, pero favorece una polinización intensa en
primavera.
● Además, otros pólenes, como los de las cupresáceas, plátano de sombra,
olivo, urticáceas o salsola, también condicionan la intensidad de la primavera para
quienes padecen alergia.
● El aumento de las enfermedades alérgicas y de los casos complejos, como la
polisensibilización, exige más alergólogos y una atención cada vez más
personalizada para los pacientes.
EL TERRITORIO ESPAÑOL
En cuanto a las gramíneas, se esperan diferencias según el territorio.
En el centro peninsular, se prevén niveles moderados a intensos en Castilla y León, Castilla-La Mancha y Madrid, con picos especialmente elevados en Toledo y Madrid, donde podrían alcanzarse concentraciones de hasta 6.000 granos por metro cúbico de aire.
En el sur peninsular, los niveles serán leves en Almería y Málaga, mientras que se situarán en valores moderados en Córdoba, Granada, Huelva y Cádiz. Las concentraciones más altas se registrarán en Badajoz y Cáceres, con valores estimados entre 10.000 y 12.000 granos/m³, así como en Sevilla, donde podrían alcanzar entre 6.000 y 8.000 granos/m³, y en Jaén, también con previsión de niveles intensos.
Por su parte, en Canarias se esperan niveles muy bajos, con concentraciones aproximadas de entre 250 y 500 granos/m³ en Tenerife y Las Palmas. En Galicia, los niveles oscilarán entre leves y moderados, especialmente en Ourense y Lugo.
En el norte peninsular —Asturias, Cantabria, País Vasco, Navarra y Aragón— se prevén niveles leves, con concentraciones estimadas entre 1.500 y 2.000 granos/m³.
Finalmente, en el litoral mediterráneo, que incluye Baleares, Cataluña, Comunidad Valenciana y Murcia, se esperan niveles leves de polen durante este periodo.
La alergia al polen ya no se limita a la primavera: los síntomas son cada vez más intensos y duraderos. El cambio climático y la contaminación están transformando el patrón tradicional de las alergias al polen. Las temporadas de polinización ya no se limitan a la primavera: comienzan antes y se prolongan durante más meses debido al aumento de las temperaturas y a los cambios en los patrones meteorológicos, que adelantan la floración y prolongan la producción de polen.
Evolución de la inmunoterapia específica con alérgenos
El Comité de Inmunoterapia de SEAIC publica una infografía sobre la evolución de la inmunoterapia con alérgenos pensada para informar a la población general.
El Comité de Inmunoterapia de la Sociedad Española de Alergología e Inmunología Clínica (SEAIC) comparte una infografía divulgativa sobre la evolución de la inmunoterapia específica con alérgenos, desde sus inicios históricos hasta los avances actuales en seguridad, estandarización y personalización del tratamiento.
![]() La evolución de la inmunoterapia específica con alérgenos SEAIC (240 descargas )
La evolución de la inmunoterapia específica con alérgenos SEAIC (240 descargas )

Captador de la semana: Sevilla – Virgen Macarena

En esta ocasión contamos con la participación de la Dra. Pilar Lara de la Rosa, alergóloga del Hospital Universitario Virgen Macarena de Sevilla, que ha preparado un vídeo muy interesante para acercarnos al captador de pólenes de Sevilla y al excelente trabajo que realizan para mantenernos informados diariamente sobre los niveles de polen a través de www.polenes.com.
ESTOY DIAGNOSTICADO DE ALFATRIPTASEMIA FAMILIAR ¿TENGO MÁS RIESGO DE TENER ALERGIA A MEDICAMENTOS?
¿Qué es la anafilaxia?
La anafilaxia es una reacción alérgica grave, que aparece de forma rápida y puede afectar a varios órganos del cuerpo (piel, aparato respiratorio, aparato digestivo y aparato cardiovascular) y requiere una atención médica urgente.
¿Qué es la triptasa?
La triptasa es una sustancia que se encuentra dentro de unas células llamadas mastocitos. Estas células participan en las reacciones alérgicas y liberan diferentes sustancias cuando se activan.
¿Qué es la alfatriptasemia familiar?
La alfatriptasemia es una condición genética descrita por primera vez en 2016. Las personas que la padecen suelen presentar niveles aumentados de triptasa en sangre, aunque en ocasiones, pueden tener valores normales. Se calcula que aproximadamente un 6% de la población puede tener esta condición, y es algo más frecuente en mujeres que en hombres.
¿Qué síntomas puede producir?
Los síntomas pueden ser muy variables. Algunas personas pueden presentar:
· Reacciones alérgicas graves (anafilaxia).
· Enrojecimiento o picor en la piel.
· Inflamación de diferentes partes del cuerpo.
· Molestias digestivas (dolor abdominal, naúseas, diarrea).
· Con menos frecuencia, pueden presentar síntomas respiratorios como asma o rinitis.
· Incluso hay pacientes que no presentan ningún síntoma.
¿Las personas con alfatriptasemia tienen más riesgo de anafilaxia?
Algunos estudios sugieren que la alfatriptasemia podría aumentar el riesgo de sufrir reacciones alérgicas graves. Por este motivo, algunos especialistas recomiendan estudiar esta condición en personas que han presentado una anafilaxia y tienen niveles a de triptasa elevados o incluso con valores considerado normales pero superiores a 8 ng/ml. No obstante, se necesitan más investigaciones para conocer con exactitud esta relación.
¿Existe más riesgo de alergia a medicamentos?
Algunos estudios han analizado si la alfa triptasemia aumenta el riesgo de reacciones alérgicas a medicamentos. Los resultados sugieren que:
· Las personas con triptasa elevada pueden presentar con más frecuencia reacciones graves tras tomar ciertos fármacos.
· Entre los pacientes que han tenido anafilaxia, entre un 13-15% tienen alfatriptasemia.
· Este hallazgo parece ser más frecuente en reacciones producidas por antibióticos y anticuerpos monoclonales.
En resumen:
· Las personas con alfatriptasemia podría tener un mayor riesgo de presentar reacciones alérgicas graves a algunos medicamentos.
· Sin embargo, la evidencia científica todavía es limitada y se necesitan más estudios para confirmarlo con seguridad.
· Si tiene alfatriptasemia y ha presentado una reacción alérgica, es recomendable comentarlo con el especialista alergólogo para valorar el caso.
Por tanto, los pacientes con alfatriptasemia tienen mayor riesgo de presentar reacciones alérgicas graves después de tomar fármacos. No obstante, harían falta estudios con más pacientes para confirmar estos datos.
Leticia Sánchez Morillas
Especialista en Alergología
Hospital Clínico San Carlos, Madrid.
EL MÚSCULO ESQUELÉTICO COMO ÓRGANO ENDOCRINO
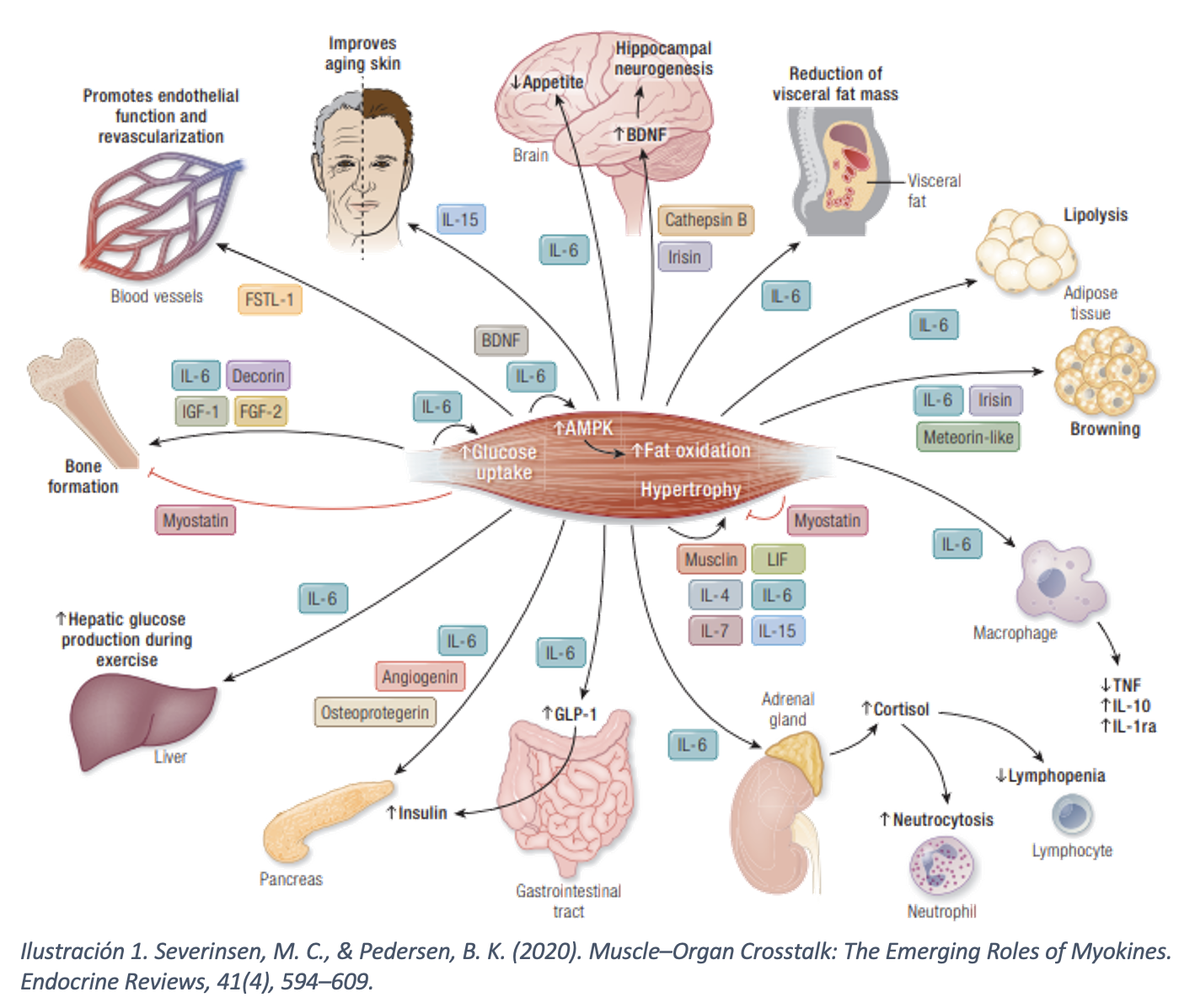
El músculo esquelético ha sido tradicionalmente reconocido por su papel considerado exclusivamente mecánico. Sin embargo, el paradigma actual lo posiciona como órgano endocrino que produce su acción mediante la secreción citoquinas que se han denominado mioquinas. Las mioquinas se producen y se liberan junto con otros péptidos por fibras musculares de forma para/auto/endocrina. Son las moléculas responsables de comunicación músculo-órganos. Gracias a ellas, el músculo modula la homeostasis metabólica y el sistema inmunitario. Una de las citoquinas más conocidas en este ámbito es la IL-6. La IL-6 en una interleucina con una función dual; modula la diferenciación de los linfocitos naive, pudiendo tener implicaciones en algunas enfermedades alergológicas como el asma, la urticaria o la dermatitis atópica.
SEÑALIZACIÓN POR LA VÍA CLÁSICA
La IL-6 que se produce durante el ejercicio señaliza predominantemente por la denominada “vía clásica”, actuando como una “hormona de esfuerzo” y ejerciendo funciones antiinflamatorias. Esta vía se activa cuando la IL-6 se une a su receptor de membrana (mIL-6R) en la célula objetivo. El receptor de IL-6 en la membrana celular está compuesto por dos subunidades:
- Receptor de IL-6 (IL-6R o CD126): Subunidad que se une a la IL-6 y la glicoproteína 130 (gp130): Subunidad transductora de señal que es esencial para que el efecto biológico de la IL-6 se lleve a cabo, formando un complejo IL-6-mIL-6R (en célula transmisora)
- Proteína gp130 (en célula receptora vecina).
De esta forma la IL6 es capaz de señalizar a las células que expresan el Receptor de Membrana (mIL-6R), entre los que se encuentran fundamentalmente:
- Hepatocitos: producción proteínas de fase aguda (reparación muscular)
- Linfocitos B: estimula su diferenciación en células plasmáticas y aumenta producción de inmunoglobulinas.
- Linfocitos T: induce respuestas antiinflamatorias a través de la producción de IL-1rα e IL-1.
- Neutrófilos y Macrófagos: ven modulada su activación por citoquinas antiinflamatorias
La IL6 señalizando por esta vía, realiza una función sistémica protectora (ej. inducción de proteínas de fase aguda en el hígado para reparación muscular) opera en condiciones dónde su concentración es baja o moderada, por lo que la IL-6 producida actúa localmente o activas células con alta expresión de mIL-6R (activando la señalización por la vía clásica). Además, existe un sistema tampón o “buffer” formado la proteína soluble gp130 (sgp130) que se escinde de las membranas celulares, en el plasma sanguíneo. En condiciones no inflamatorias, el sistema tampón permite la señalización homeostática local principalmente en células inmunitarias y hepatocitos, pero neutraliza la IL6 antes de que pueda activar su vía patológica (neutralizando el complejo IL6-sIL6R) evitando que se activen otras células como las células musculares lisas y células endoteliales, que no son las células con alta expresión de receptor de membrana de IL-6. Este mecanismo funciona porque el complejo IL6-sIL6R se une con mayor afinidad a la versión soluble de la proteína gp130, que a su versión de membrana, al unirse el sgp130, el complejo IL-6-sIL6R-sgp130 queda inactivado y secuestrado en el plasma, impidiendo que la IL-6 provoque efectos proinflamatorios a través de la señalización patológica o trans-señalización
Un nivel bajo de IL-6 y el sistema tampón previenen la señalización sistémica patológica de IL-6. En condiciones inflamatorias agudas y crónicas, el sistema tampón se sobrecarga y permite la trans-señalización sistémica de IL-6.
SEÑALIZACIÓN POR LA VÍA “TRANS”
La obesidad crónica causa que los adipocitos hipertrofiados se vuelvan disfuncionales y entren en apoptosis. Esto atrae a los macrófagos para eliminarlos. Estos macrófagos proinflamatorios forman estructuras llamadas «coronas» alrededor de estos adipocitos. Estos macrófagos liberan citoquinas altamente proinflamatorias, que incluyen IL-6, y también TNF−α e IL−1β. La IL-6 en estas circunstancias amplifica la señalización proinflamatoria. El tejido adiposo inflamado y los macrófagos liberan grandes cantidades del receptor soluble de IL-6 (sIL-6R) a la circulación. Al haber gran concentración de sIL-6R, la IL-6 se une a él y forma un complejo (sIL-6R-IL-6) que activa la señalización por la vía trans. Esta vía activa de forma patológica las células que normalmente no responden a la IL-6 (como las células endoteliales y las células musculares), generando entre otros efectos crónicos, resistencia a la insulina y disfunción endotelial. En estas circunstancias, la liberación de IL-6 visceral en la obesidad y en otros transtornos inflamatorios, es crónica y sostenida, agotando el mecanismo de neutralización (buffer).
IL-6 Y EJERCICIO FÍSICO
El músculo esquelético puede aumentar la concentración de IL-6 plasmática hasta x1000 durante el ejercicio. La magnitud de esta respuesta viene determinada por las características del ejercicio. Esto ocurre porque disponemos de dos tipos de fibras musculares:
- Fibras Tipo I: Fibras de contracción lenta, oxidativas, resistentes a la fatiga.
- Fibras Tipo II: fibras de contracción rápida, glicolíticas, utilizadas para fuerza y potencia.
Duración e intensidad del ejercicio
La duración del ejercicio se relaciona de forma directa con la concentración de IL-6 muscular, lo que supone el estímulo más importante para el aumento de IL-6 plasmática. La concentración de IL-6 dentro de las fibras musculares aumenta más que la concentración plasmática, ya que el músculo actúa como reservorio. Las fibras musculares de tipo I son la principal fuente de IL6 plasmático. Este tipo muscular es el tipo reclutado en ejercicio de larga duración y moderada intensidad. La concentración de IL-6 es proporcional a la duración del ejercicio, aumentado de forma exponencial. También cae exponencialmente poco después de finalizar
La producción de IL6 se amplifica en ejercicio de mayor intensidad ante igual duración. Aparentemente se requiere un mínimo de intensidad para inducir la respuesta de I-L6, al menos en las actividades de baja duración.
La interacción tanto de la intensidad como de la duración del ejercicio son importantes para determinar la respuesta de IL6 plasmática con respecto al ejercicio
TIPO DE EJERCICIO Y ESTADO DE ENTRENAMIENTO
Tipo de ejercicio: Estudios longitudinales sugieren que las personas entrenadas pueden producir más cantidad de IL-6 que las no entrenadas incluso cuando trabajan a la misma intensidad. Este efecto es menor en comparación con intensidad y duración del ejercicio en general. Algunos tipos de ejercicio producen concentraciones plasmáticas superiores a otros (p. ej: más IL-6 plasmática en carrera Vs bicicleta, probablemente debido a que se produce contracción excéntrica y más daño muscular en la carrera, por lo que se produce fuga de IL-6 a la circulación, y reclutamiento de leucocitos que también aumentan la producción de IL-6).
Estado de entrenamiento: La concentración de IL-6 en reposo no reflejan la liberación muscular sino la producción por otras vías, inflamatorias a través de leucocitos, adipocitos disfuncionales y su acumulación, por lo que es indicativa del estado de inflamación del individuo, de hecho es inversamente proporcional a la actividad física habitual. Los programas de entrenamiento han demostrado disminuir la IL-6 en reposo de población inactiva o con inflamación crónica, acompañado de la disminución de otros marcadores inflamatorios (PCR, TNFα). Además, en atletas de élite con concentraciones de IL-6 plasmáticas muy inferiores a población con enfermedades crónicas, aumentando la cantidad de entrenamiento no se produce mayor disminución de concentración de IL-6 con respecto a sus valores en reposo, por lo que es posible que los atletas de élite con altas cargas de entrenamiento, no puedan disminuir AUN MÁS su concentración de IL-6.
Además, se ha sugerido que el sobre-entrenamineto puede producir un aumento de la concentración de IL-6 basal y un estado inflamatorio. Se ha observado este efecto en personas con periodos cortos de entrenamiento de muy alta intensidad y en deportistas con sobre-entrenamiento.
Se hipotetiza entonces que la curva que relaciona la concentración de IL-6 y carga de entrenamiento tiene forma de U, tanto en individuos sedentarios como en entrenados. De esta forma, encontramos valores altos de IL-6 en reposo en pacientes sedentarios debido al estado inflamatorio, y en deportista sobreentrenados, encontrando menores niveles de IL-6 basal en pacientes con altas cargas de entrenamiento, que no han llegado al sobre-entrenamineto.
En este sentido, en estudios experimentales en ratones se ha observado que Tozilizumab deteriora el aumento de la masa del VI tras 12 semanas de entrenamiento aeróbico en comparación con placebo, y que las infusiones de IL6 recombinante junto con un programa de ejercicio, condujeron en modelos murinos, a un aumento del rendimiento en comparación con el mismo programa sin IL6.
Por este motivo, hay que entender el músculo esquelético como un órgano endocrino dinámico, capaz de modular el estado inflamatorio sistémico a través de la secreción de IL-6. La clave de su impacto no reside en la molécula en sí, sino en el contexto de su señalización. Mientras que la obesidad y el estado proinflamatorio saturan el mecanismo de neutralización de la IL-6 (buffer de sgp130), permitiendo una trans-señalización patológica que daña el endotelio y el metabolismo, el ejercicio físico favorece la señalización por la vía clásica, transformando a la IL-6 en una hormona de esfuerzo esencial para la reparación muscular y efecto antiinflamatorio.
El ejercicio regular de intensidad moderada actúa como el estímulo idóneo para favorecer la inmunomodulación en la inflamación crónica, subrayando que la IL-6 muscular no es solo un subproducto del esfuerzo, sino una señal biológica indispensable para la homeostasis del organismo.
INMUNOMODULACIÓN VÍA IL-6
La IL-6 desempeña un papel crítico en la modulación del sistema inmune adaptativo. En los linfocitos B es esencial para la diferenciación hacia cél. plasmáticas productoras de inmunoglobulinas. En los linfoctios T influye en el equilibrio de subpoblaciones, promoviendo la diferenciación de los linfocitos T naive hacia Th17, y limitando la diferenciación hacia linfocitos T reguladores al limitar la expresión Foxp3 (interruptor genético Treg)
Al promover TH17 e inhibir Treg, desplaza el equilibrio inmune hacia un estado pro-inflamatorio y autoinmune. Los Treg actúan para suprimir la actividad de todas las células T efectoras (IL10 suprime Th1, Th2 y Th17), manteniendo así la homeostasis y evitando respuestas inmunes inapropiadas. Si los T reguladores funcionan adecuadamente suprime la actividad de las células T efectoras, incluyendo T2, favoreciendo la tolerancia a los alérgenos, reduciendo la intensidad de la respuesta alérgica y la producción de IgE. Por este motivo el ejercicio moderado y crónico disminuye de forma indirecta respuesta T2 promoviendo un ambiente antiinflamatorio sistémico.
En enfermedades como el asma o la dermatitis atópica grave, la IL-6 actúa como un puente entre la inflamación T2 y la inflamación Th17 donde la liberación de IL6 e IL−1β (ambiente inflamatorio) favorecen la supresión T reguladoras y la diferenciación hacia Th17 con una inflamación predominantemente neutrofilica que se añade a la inflamación T2 preexistente, resultando en un aumento de complejidad de la enfermedad y confiriendo resistencia a tratamiento con T2 como diana. En algunos estudios, por este motivo, se propone IL6 como biomarcador de un endotipo de asma no-T2.
De forma paralela se ha observado un desequilibrio Th17/Treg en la urticaria crónica espontanea mal controlada, por lo que parece ser un marcador relevante de la gravedad UCE, (asociándose a más prurito, peor calidad de vida y mayor actividad de la enfermedad), encontrando que el ejercicio a corto plazo mejora los síntomas, mientras los programas de ejercicio regular a largo plazo reducen los síntomas y mejoran las puntuaciones UCT y la calidad de vida.
Estos paralelismos también los encontramos en otras enfermedades atópicas como la dermatitis atópica (DA): En células mesenquimales de la piel de pacientes con dermatitis atópica se ha observado un perfil inflamatorio alterado en comparación con pacientes sanos, con sobreexposición de IL6 de forma significativa, y en modelos murinos con eccema tipo DA el ejercicio aeróbico de intensidad moderada atenúa los síntomas de DA modulando los mediadores inflamatorios. Estos resultados sugieren que el ejercicio puede ser una terapia complementaria eficaz y práctica en DA al reducir los mediadores inflamatorios.
CONCLUSIONES
- El músculo esquelético debe ser considerado un órgano endocrino.
- Se comunica con el resto de órganos y células mediante la secreción de mioquinas
- La IL-6 es una de las mioquinas más descritas. La función que ejerce depende el contexto
- El ejercicio actúa como un modulador inmunológico al influir en la IL-6 y por tanto linfocitos T y B
- El ejercicio regular puede ayudar a mitigar la disregulación inmunitaria característica de las enfermedades alérgicas y autoinmunes al modular el eje Treg/TH17 y reducir la inflamación crónica basal.
- La prescripción de ejercicio debe verse como una intervención terapéutica complementaria en el manejo de enfermedades alérgicas e inflamatorias.
- No todo el ejercicio es igual: Para lograr el efecto antiinflamatorio sistémico y la modulación inmune óptima, la duración, intensidad y tipo de actividad física son consideraciones clave para futuras investigaciones y recomendaciones clínicas.
BIBLIOGRAFÍA
- Severinsen, M. C., & Pedersen, B. K. (2020). Muscle–Organ Crosstalk: The Emerging Roles of Myokines. Endocrine Reviews, 41(4), 594–609.
- Nash D, Hughes MG, Butcher L, Aicheler R, Smith P, Cullen T, Webb R. IL-6 signaling in acute exercise and chronic training: Potential consequences for health and athletic performance. Scand J Med Sci Sports. 2023 Jan;33(1):4-19.
- Rose-John S, Jenkins BJ, Garbers C, Moll JM, Scheller J. Targeting IL-6 trans-signalling: past, present and future prospects. Nat Rev Immunol. 2023 Oct;23(10):666-681. doi: 10.1038/s41577-023-00856-y
- Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014 Sep 4;6(10):a016295. doi: 10.1101/cshperspect.a016295. PMID: 25190079
- Luan, D., Dadpey, B., Zaid, J., Bridge-Comer, P., DeLuca, J., Xia, W., … & Reilly, S. (2022). Adipocyte-secreted il-6 sensitizes macrophages to il-4 signaling.. https://doi.org/10.1101/2022.07.19.500620
- Namakanova, O. A., Горшкова, Е. А., Зварцев, Р. В., Nedospasov, S. A., Drutskaya, M. S., & Gubernatorova, E. O. (2022). Therapeutic potential of combining il-6 and tnf blockade in a mouse model of allergic asthma. International Journal of Molecular Sciences, 23(7), 3521.
- Rose-John S, Jenkins BJ, Garbers C, Moll JM, Scheller J. Targeting IL-6 trans-signalling: past, present and future prospects. Nat Rev Immunol. 2023 Oct;23(10):666-681. doi: 10.1038/s41577-023-00856-y
- Gupta, D., Orehek, S., Turunen, J. J., O’Donovan, L., Gait, M. J., Andaloussi, S. E., … & Wood, M. J. A. (2023). Modulation of pro-inflammatory il-6 trans-signaling axis by splice switching oligonucleotides as a therapeutic modality in inflammation. Cells, 12(18), 2285.
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003 Dec 15;198(12):1875-86
- Lee J, Lozano-Ruiz B, Yang FM, Fan DD, Shen L and Gonzá lezNavajas JM (2021) The Multifaceted Role of Th1, Th9, and Th17 Cells in Immune Checkpoint Inhibition Therapy. Front. Immunol. 12:625667
- Ryba‐Stanisławowska, M., Skrzypkowska, M., Myśliwska, J., & Myśliwiec, M. (2013). The serum il-6 profile and treg/th17 peripheral cell populations in patients with type 1 diabetes. Mediators of Inflammation, 2013, 1-7.
- Kong et al. «Antigen‐specific transforming growth factor β–induced Treg cells, but not natural Treg cells, ameliorate autoimmune arthritis in mice by shifting the Th17/Treg cell balance from Th17 predominance to Treg cell predominance» Arthritis & rheumatism
- Huss et al. «TGF-β Enhances Effector Th1 Cell Activation but Promotes Self-Regulation via IL-10“ The Journal of Immunology, vol. 184, no. 8, 2010.
- Palomares, O., et al. (2010). T regulatory cells and allergic disease. European Journal of Immunology, 40(5), 1266–1271.
- Lindsley AW, Lugogo N, Reeh KAG, Spahn J, Parnes JR. Asthma Biologics Across the T2 Spectrum of Inflammation in Severe Asthma: Biomarkers and Mechanism of Action. J Asthma Allergy. 2025 Jan 14;18:33-57. doi: 10.2147/JAA.S496630. PMID: 39830595; PMCID: PMC11742565.
- Son WK, Yoon W, Kim S, Byeon JH, Lee JS, Kim D, Jaehoon L, Chae Y, Yoon SJ, Yoo Y. Can moderate-intensity aerobic exercise ameliorate atopic dermatitis? Exp Dermatol. 2020 Aug;29(8):699-702. doi: 10.1111/exd.14138. Epub 2020 Jul 20. PMID: 32614478.
- Campanati A, Orciani M, Marani A, Di Vincenzo M, Magi S, Gregoriou S, Diotallevi F, Martina E, Radi G, Offidani A. Mesenchymal Stem Cells Profile in Adult Atopic Dermatitis and Effect of IL4-IL13 Inflammatory Pathway Inhibition In Vivo: Prospective Case-Control Study. J Clin Med. 2022 Aug 15;11(16):4759. doi: 10.3390/jcm11164759. PMID: 36013001; PMCID: PMC9409772.
Si tengo una reacción con un antiinflamatorio no esteroideo (AINE), ¿tengo que evitar todos hasta ser valorado por alergología?
Los antiinflamatorios no esteroideos (AINE) son los fármacos más consumidos a nivel mundial y la primera causa de hipersensibilidad a medicamentos, cuya prevalencia continúa en aumento.
¿Qué es un AINE? El término AINE no se refiere a un único medicamento, sino a una gran variedad de moléculas (como el ibuprofeno, el diclofenaco, la Aspirina ®, etc.) que comparten un mecanismo de acción: la inhibición de las enzimas ciclooxigenasas (COX-1 y/o COX-2). Estas enzimas son proteínas muy importantes en nuestro organismo para mantener el funcionamiento normal de la mucosa del aparato digestivo, del riñón y de las plaquetas. La prescripción de un AINE se basa en su acción antiagregante de la función plaquetaria, así como en la capacidad para reducir la inflamación y el dolor, y por ello su indicación terapéutica es muy común. Se llaman así para distinguirlos de otros antiinflamatorios esteroideos que pertenecen al grupo de la cortisona. Los AINEs se clasifican en varios grupos químicos (tabla 1).
Si se presenta una reacción de hipersensibilidad tras el uso de un AINE, es habitual alarmarse y preguntarse: ¿Tengo que evitar todos hasta ser valorado por alergología? La respuesta es sí, de forma cautelar hasta tener un diagnóstico y recomendación específica por un especialista en Alergología.
¿Cuál es el motivo de esta precaución inicial tan importante y amplia? La existencia de diferentes tipos de manifestaciones clínicas que abarcan desde síntomas cutáneos (urticaria, angioedema), respiratorios (rinitis, dificultad respiratoria, sibilantes…) y generales (como hipotensión y anafilaxia) que pueden presentarse de forma aislada o conjunta y originarse tras pocos minutos de su administración o en horas tras ello. Esta complejidad es debida a la coexistencia de diferentes mecanismos que todavía no están completamente comprendidos y se traduce en la posibilidad de presentar una reacción tras el uso de un AINE estructuralmente distintos entre sí (lo más frecuente), como por ejemplo Aspirina e ibuprofeno, o de una reacción selectiva a sólo un AINE o a aquellos con una estructura química similar (menos frecuentes), como por ejemplo el Nolotil ®.
El camino a seguir: la valoración alergológica. Para así confirmar el diagnóstico, identificar si la reacción es a todos los AINEs (cruzada) o específica e indicar alternativas de antiinflamatorios seguros para su uso en el futuro.
Gádor Bogas
Especialista en Alergología
Hospital Regional Universitario de Málaga
Actualización en las Inmunodeficiencias primarias: errores innatos de la inmunidad
1.Introducción
El sistema inmunitario constituye un entramado altamente especializado de células, tejidos y mediadores solubles cuya función principal es la defensa frente a los agentes infecciosos, así como el mantenimiento de la tolerancia frente a los antígenos propios. Las alteraciones congénitas que afectan al desarrollo, diferenciación o función de cualquiera de estos componentes dan lugar a un grupo amplio y heterogéneo de enfermedades hereditarias tradicionalmente conocidas como inmunodeficiencias primarias.
En las últimas décadas, el avance en el conocimiento de la inmunología molecular y de la genética humana ha permitido identificar un número creciente de defectos monogénicos responsables de estos cuadros. Este progreso ha llevado a una redefinición conceptual, adoptándose el término errores innatos de la inmunidad (EII) , que refleja de forma más precisa la diversidad de mecanismos patogénicos implicados y el amplio espectro clínico asociado.
El concepto de EII engloba no solo las formas clásicas caracterizadas por infecciones recurrentes o graves, sino también aquellas en las que predominan fenómenos de desregulación inmunitaria, como la autoinmunidad, la autoinflamación, la alergia grave, la linfoproliferación o la predisposición a neoplasias. En muchos casos, estas manifestaciones coexisten en un mismo paciente, lo que pone de manifiesto que los EII representan modelos de disfunción inmunológica, más que simples estados de inmunodeficiencia cuantitativa. Desde un punto de vista clínico, este cambio obliga a ampliar el enfoque diagnóstico más allá del patrón infeccioso clásico y justifica la integración temprana de herramientas genéticas y moleculares en el proceso diagnóstico.
- Epidemiología y relevancia clínica de los errores innatos de la inmunidad
Los EII son considerados enfermedades raras de forma individual, pero en conjunto representan un problema de salud pública mayor de lo que se estimaba tradicionalmente. Estudios poblacionales recientes sitúan su prevalencia global entre 1 por cada 1.000 y 1 por cada 5.000 habitantes, cifras claramente superiores a las recogidas en registros históricos, lo que sugiere un infradiagnóstico significativo, especialmente de las formas leves o de inicio tardío.
La clasificación de la Unión Internacional de las Sociedades de Inmunología (IUIS) se actualiza de forma periódica y refleja el rápido crecimiento del conocimiento en este campo. En la actualización de 2024 se describen más de 500 genes implicados en EII, con más de 550 entidades clínicas reconocidas, incluyendo defectos debidos a variantes germinales, mutaciones somáticas y fenocopias mediadas por autoanticuerpos neutralizantes.
La distribución de los EII varía según el tipo de defecto, la población estudiada y el acceso a herramientas diagnósticas avanzadas. Las inmunodeficiencias predominantemente de anticuerpos continúan siendo las más frecuentes en registros clínicos, mientras que las inmunodeficiencias combinadas graves son menos prevalentes, pero con una carga de morbimortalidad mucho mayor si no se diagnostican y tratan de forma precoz, por lo que se han incluido en las pruebas de cribado neonatal.
Desde el punto de vista pediátrico, los EII constituyen un reto diagnóstico relevante. Muchas de sus manifestaciones iniciales, como las infecciones respiratorias recurrentes, pueden confundirse con procesos habituales de la infancia, especialmente en los primeros años de vida o en el contexto de la escolarización. Este solapamiento con la normalidad explica en parte el retraso diagnóstico observado en numerosos estudios, que puede alcanzar varios años incluso en países con sistemas sanitarios avanzados.
El impacto clínico del retraso diagnóstico es considerable, ya que favorece la aparición de secuelas irreversibles, como bronquiectasias, daño pulmonar crónico, retraso ponderoestatural, complicaciones autoinmunes o infecciones potencialmente mortales. Por este motivo, la concienciación de los profesionales sanitarios y el reconocimiento precoz de los signos de alarma son elementos fundamentales para mejorar el pronóstico de estos pacientes.
- Espectro clínico de los errores innatos de la inmunidad
El espectro clínico de los EII es extraordinariamente amplio y heterogéneo. Aunque históricamente se han asociado de forma predominante a infecciones recurrentes, actualmente se reconoce que estas representan solo una parte del abanico de manifestaciones posibles.
3.1 Manifestaciones infecciosas
Las infecciones continúan siendo la forma de presentación más frecuente en la infancia . Suelen caracterizarse por una frecuencia aumentada de infecciones, una mayor gravedad de estas, escasa respuesta a los tratamientos habituales o producida por microorganismos infrecuentes u oportunistas. El tipo de infección puede orientar hacia el compartimento inmunológico afectado. Así, las infecciones por bacterias encapsuladas sugieren defectos de anticuerpos o del complemento, mientras que las infecciones virales persistentes, micobacterias u hongos son más características de inmunodeficiencias combinadas o de los defectos de la inmunidad celular (Tabla 1)
Tabla 1. Clasificación de los Errores innatos de la inmunidad
- Inmunodeficiencias combinadas graves
- Inmunodeficiencias combinadas más leves que asocian características sindrómicas
- Inmunodeficiencias predominantemente de anticuerpos
- Enfermedades por desregulación inmune
- Defectos del número y función de las células fagocíticas
- Defectos de la inmunidad intrínseca e innata
- Enfermedades autoinflamatorias
- Defectos de la cascada del complemento
- Fallos de médula
- Fenocopias de inmunodeficiencias congénitas
3.2 Manifestaciones no infecciosas
Un porcentaje importante de los pacientes con EII presenta manifestaciones no infecciosas como forma predominante o incluso como forma única de debut. Entre ellas destacan:
- Citopenias autoinmunes
- Enfermedad inflamatoria intestinal
- Dermatitis grave de inicio precoz
- Fiebre recurrente sin causa infecciosa
- Linfadenopatías persistentes
- Hepatoesplenomegalia
- Fenómenos granulomatosos
- Neoplasias hematológicas.
Estas presentaciones pueden retrasar el diagnóstico si no se consideran dentro del espectro de los EII.
- Signos de alerta y sospecha clínica en pediatría
Con el objetivo de facilitar el reconocimiento precoz de los EII, se han propuesto diversos sistemas de cribado clínico. Los diez signos de alerta clásicos continúan siendo una herramienta útil en la práctica clínica, especialmente en Atención Primaria, aunque deben interpretarse de forma flexible y contextualizada. De forma resumida, deben hacer sospechar un EII:
- Infecciones recurrentes, graves o por microrganismos poco frecuentes
- Necesidad repetida de antibióticos intravenosos
- Fracaso del tratamiento antibiótico convencional
- Fallo de medro o retraso del crecimiento
- Antecedentes familiares de EII
- Reacciones adversas a vacunas por microrganismos vivos
- Manifestaciones autoinmunes o inflamatorias persistentes y de debut precoz
- Bronquiectasias sin causa aparente
- Diarrea crónica
Es especialmente importante insistir en que la ausencia de infecciones recurrentes no excluye un EII, y que la coexistencia de infecciones con autoinmunidad, inflamación o alergia grave debe aumentar de forma significativa el nivel de sospecha clínica.
- Clasificación de los errores innatos de la inmunidad
El conocimiento de los EII ha experimentado una expansión extraordinaria en las últimas dos décadas, impulsada fundamentalmente por el desarrollo de técnicas de secuenciación masiva y por la caracterización de nuevas vías inmunológicas. Este avance exige una actualización periódica de su clasificación, con el objetivo de facilitar la orientación diagnóstica y terapéutica.
La clasificación de la Unión Internacional de las Sociedades de Inmunología (IUIS) constituye el marco de referencia internacional para la organización de los EII. La actualización más reciente (2024) reconoce más de 500 defectos genéticos responsables de EII humanos, organizados en 10 grandes grupos según el fenotipo clínico e inmunológico predominante. Es importante destacar que esta clasificación no es rígida, ya que un mismo defecto genético puede dar lugar a manifestaciones clínicas incluidas en diferentes categorías.
A diferencia de clasificaciones previas, el modelo actual incorpora de forma explícita conceptos clave como variantes de ganancia de función (GOF) y pérdida de función (LOF), mutaciones somáticas, fenocopias mediadas por autoanticuerpos y mecanismos dominantes negativos o haploinsuficiencia.
5.1 Inmunodeficiencias combinadas
Las inmunodeficiencias combinadas se caracterizan por una alteración significativa de la inmunidad celular mediada por linfocitos T, con un compromiso variable de la inmunidad humoral. Constituyen uno de los grupos más graves dentro de los EII, especialmente cuando el déficit de linfocitos T es profundo. Las formas más graves se agrupan bajo el término inmunodeficiencia combinada grave (SCID), que suele manifestarse en los primeros meses de vida con infecciones graves, recurrentes u oportunistas, diarrea crónica, fallo de medro y, en ocasiones, lesiones cutáneas compatibles con enfermedad injerto contra huésped secundaria al quimerismo materno.
Los patógenos implicados incluyen bacterias, virus, hongos y protozoos oportunistas. Desde el punto de vista inmunológico, estas entidades se caracterizan por linfopenia T muy marcada, con afectación variable de linfocitos B y células NK, lo que da lugar a los distintos fenotipos inmunológicos clásicos. La identificación precoz de estas formas es crucial, ya que constituyen una urgencia pediátrica, y su pronóstico depende en gran medida de la rapidez con la que se instaura las medidas preventivas y el tratamiento curativo, habitualmente el trasplante de progenitores hematopoyéticos y en ocasiones la terapia génica, de ahí la gran relevancia que tiene su reciente implementación en la pruebas en el cribado neonatal.
5.2 Inmunodeficiencias combinadas con características sindrómicas
Este grupo incluye EII en los que la alteración inmunitaria se acompaña de anomalías congénitas o rasgos dismórficos que afectan a otros órganos o sistemas. Ejemplos clásicos son los síndromes asociados a alteraciones del desarrollo tímico, defectos de la reparación del ADN o trastornos del citoesqueleto. Clínicamente, estos pacientes pueden presentar infecciones recurrentes junto con malformaciones craneofaciales, cardiopatías congénitas, retraso del desarrollo psicomotor, alteraciones cutáneas o anomalías esqueléticas. La gravedad del compromiso inmunitario es variable, oscilando desde formas leves hasta inmunodeficiencias combinadas graves. La presencia de rasgos sindrómicos puede facilitar el reconocimiento clínico del EII, pero también puede desviar la atención hacia el defecto estructural y retrasar la valoración inmunológica, por lo que es fundamental mantener un enfoque integral.
5.3 Inmunodeficiencias predominantemente de anticuerpos
Las inmunodeficiencias predominantemente de anticuerpos constituyen el grupo más frecuente de EII en la práctica clínica, representando la mayoría de los casos diagnosticados tanto en la infancia como en la edad adulta. Se caracterizan por una producción deficiente de inmunoglobulinas, una alteración en la respuesta específica frente a antígenos o ambas. Clínicamente, se manifiestan de forma típica a partir de los primeros 5 meses de vida, una vez que desaparece protección conferida por las inmunoglobulinas maternas, aunque algunas entidades pueden debutar más tardíamente.
Las manifestaciones clínicas predominantes son las infecciones del tracto respiratorio y gastrointestinal recurrentes, producidas principalmente por bacterias encapsuladas. En pacientes de mayor edad pueden aparecer complicaciones como bronquiectasias, enfermedad pulmonar crónica, linfoproliferación y un aumento del riesgo de enfermedades autoinmunes y neoplasias, especialmente hematológicas. Es importante destacar que, en este grupo, las manifestaciones no infecciosas, especialmente las citopenias autoinmunes y la enfermedad inflamatoria intestinal, pueden preceder al diagnóstico inmunológico, lo que refuerza la necesidad de considerar un EII ante cuadros autoinmunes de inicio precoz o de evolución atípica o tórpida.
5.4 Enfermedades de desregulación inmunitaria
Las enfermedades de desregulación inmunitaria constituyen un grupo cada vez más relevante dentro de los EII. En estas entidades, el defecto primario no es tanto la incapacidad para defenderse frente a infecciones como la pérdida del control de la respuesta inmunitaria. Desde el punto de vista clínico, se caracterizan por debutar con autoinmunidad a edades muy temprana y en formas muy graves, inflamación crónica, linfoproliferación, fiebre recurrente y, en algunos casos, susceptibilidad a infecciones. La afectación puede ser multisistémica y de curso fluctuante. El reconocimiento de estos EII ha tenido importantes implicaciones terapéuticas, ya que muchas de estas entidades responden a tratamientos dirigidos contra vías inmunológicas específicas, como inhibidores de la Janus Kinasas (JAK) o terapias biológicas, lo que ha modificado de forma sustancial su pronóstico.
5.5 Defectos congénitos de fagocitos
Los defectos congénitos del número o la función de los fagocitos incluyen entidades en las que existe una alteración de la quimiotaxis, o de la fagocitosis o de la capacidad microbicida de neutrófilos y macrófagos. Clínicamente, se caracterizan por infecciones bacterianas y fúngicas recurrentes, a menudo con formación de abscesos en órganos internos, adenitis supurativas o infecciones por microorganismos poco habituales. En algunas entidades se observan fenómenos inflamatorios crónicos y formación de granulomas, que pueden afectar a diferentes órganos. La edad de presentación es variable y depende del grado de afectación funcional. La identificación de este grupo es fundamental para instaurar profilaxis antimicrobiana específica y valorar tratamientos inmunomoduladores o curativos.
5.6 Defectos de la inmunidad innata e intrínseca
Este grupo incluye EII en los que existe una alteración selectiva de mecanismos de la inmunidad innata, responsables del reconocimiento temprano de patógenos y de la activación de la respuesta inmunitaria adaptativa. A diferencia de otros EII, estos defectos suelen manifestarse por susceptibilidad a infecciones muy concretas, como infecciones virales graves, infecciones por micobacterias o infecciones invasivas por bacterias encapsuladas, mientras que el resto de la respuesta inmunitaria puede ser aparentemente normal. El reconocimiento de estos patrones clínicos específicos es clave para orientar el diagnóstico, ya que las pruebas inmunológicas convencionales pueden ser normales.
5.7 Enfermedades autoinflamatorias
Las enfermedades autoinflamatorias se caracterizan por episodios recurrentes de inflamación sistémica no mediada por autoanticuerpos ni linfocitos autorreactivos, sino por una activación inapropiada de la inmunidad innata, aunque también puede estar afectada la inmunidad adaptativa. En la infancia, suelen manifestarse con fiebre recurrente, elevación de reactantes de fase aguda, afectación cutánea, articular o serosa, y un curso clínico fluctuante. Aunque clásicamente se han considerado entidades diferenciadas, actualmente se incluyen dentro del espectro de los EII, dada su base genética y su solapamiento con otras formas de desregulación inmunitaria. Hay un protocolo especifico de enfermedades autoinflamatorias donde se tratan las mismas con más profundidad.
5.8 Deficiencias del complemento
Las deficiencias del sistema del complemento representan un grupo relativamente infrecuente de EII, pero con manifestaciones clínicas características. Se asocian principalmente a infecciones recurrentes por bacterias encapsuladas, en especial Neisseria spp., así como a enfermedades autoinmunes mediadas por inmunocomplejos. La sospecha clínica debe plantearse ante episodios repetidos de meningitis o sepsis meningocócica, así como en pacientes con lupus eritematoso sistémico de inicio precoz. Es importante recordar que déficit de C1 inhibidor provoca angioedema hereditario, caracterizado por edemas recurrentes no pruriginosos, sin urticaria y con mala respuesta a antihistamínicos o corticoides. Por otro lado, las alteraciones de la vía alternativa del complemento factor H, I, MCP, C3) se asocian al síndrome hemolítico urémico atípico, con microangiopatía trombótica, insuficiencia renal y alto riesgo de recurrencia.
5.9 Fallo medular
Las entidades incluidas en este grupo se caracterizan por una alteración primaria de la hematopoyesis, que puede asociarse a inmunodeficiencia, infecciones recurrentes, citopenias y predisposición a malignidad. La afectación inmunológica puede pasar desapercibida inicialmente, predominando las manifestaciones hematológicas, lo que subraya la importancia de una evaluación inmunológica completa en niños con fallo medular de causa no filiada.
5.10 Fenocopias de errores innatos de la inmunidad
El último grupo incluye entidades que clínicamente simulan un EII monogénico clásico, pero cuya causa no es una mutación germinal heredada. Se incluyen aquí mutaciones somáticas, autoanticuerpos neutralizantes frente a citocinas o mediadores inmunes y otros mecanismos adquiridos. El reconocimiento de estas fenocopias es esencial, ya que su manejo y pronóstico pueden diferir de los EII congénitos .
- Inmunodeficiencias y Atopia: Trastornos Atópicos Primarios (PADs)
Las inmunodeficiencias primarias con fenotipo alérgico se agrupan bajo el concepto de Trastornos Atópicos Primarios (Primary Atopic Disorders, PADs), definidos como enfermedades monogénicas en las que la atopia grave, precoz y multifacética (eccema, asma, alergias alimentarias, eosinofilia) es una manifestación central, frecuentemente asociada a inmunodeficiencia y desregulación inmune. Se han identificado más de 48 defectos genéticos responsables de PADs, que incluyen alteraciones en la señalización de citocinas, función de células T reguladoras, diferenciación Th2/Th17, y defectos de la barrera epitelial.
Ejemplos son el síndrome de hiper-IgE (STAT3 dominante, DOCK8 recesivo), caracterizado por eccema grave, infecciones recurrentes, IgE elevada y eosinofilia; el síndrome de Omenn (mutaciones en RAG1/2), con eritrodermia, eosinofilia, IgE elevada y expansión clonal de linfocitos T; y la reciente descripción de variantes de ganancia de función en STAT6, que causan dermatitis atópica grave, hipereosinofilia, asma, alergias alimentarias y anafilaxia, con herencia autosómica dominante o casos esporádicos. En estos pacientes, la hiperactivación sostenida de STAT6 induce polarización Th2 y sobreexpresión de genes diana alérgicos, y se ha demostrado eficacia clínica con dupilumab y con inhibidores JAK-STAT como ruxolitinib.
La distinción entre EII monogénicas y atopía poligénica es fundamental: la atopía poligénica suele ser multifactorial y de inicio tardío, mientras que los PADs se presentan con atopia grave, precoz, multiorgánica y frecuentemente asociada a infecciones recurrentes, autoinmunidad o linfoproliferación. Las «banderas rojas» clínicas que deben alertar al clínico incluyen: atopia grave y resistente al tratamiento, inicio en la primera infancia, coexistencia de múltiples manifestaciones alérgicas, infecciones recurrentes, eosinofilia marcada, IgE extremadamente elevada, historia familiar sugestiva y presencia de autoinmunidad o linfoproliferación.
6.1 Características comunes de la IDPs con fenotipos atópicos.
- Enfermedad atópica de inicio temprano, generalmente al nacer o en los primeros meses de vida.
- Enfermedad atópica grave, que generalmente no responde a la terapia estándar (por ejemplo, eccema grave recalcitrante)
- Niveles altos de biomarcadores T2 (por ejemplo, aumento de IgE sérica total, eosinofilia)
- Presencia de otros miembros de la familia afectados (patrón de herencia, incluidos antecedentes familiares de inmunodeficiencias primarias y / o diátesis atópica grave familiar), antecedentes familiares de consanguinidad
- Asociación de atopia con infecciones recurrentes/graves (especialmente por patógenos oportunistas o herpes virus
- Presencia de autoinmunidad, linfopenia o citopenias asociadas al cuadro alérgico
No podemos olvidar que en muchas ocasiones los EII pueden presentar dermatitis urticarias, rinitis y asma de etiología alergica, así como cuadro alergia alimentarias, si bien estos cuadros suelen ser más graves que en la población alérgica en la que no hay una IDP de base, rasgo en sí que aunque no está incluido como signo de alarma clásico si suele ser un constante
Para el alergólogo, es crucial identificar los PADs, enfermedades monogénicas donde la atopia grave y precoz es la manifestación central. La identificación de un EII permite transitar hacia una medicina de precisión. El alergólogo debe mantener una sospecha activa ante atopias «atípicas», ya que los síntomas alérgicos pueden ser el epifenómeno de un defecto molecular profundo que compromete la supervivencia del paciente
Bibliografía
- Poli MC, et al. Human inborn errors of immunity: 2024 update on the classification (IUIS). J Hum Immun. 2025.
- Ameri S, et al. Primary immunodeficiency diseases: updated practice parameter. J Allergy Clin Immunol. 2022.
- Kindle G, et al. Inborn errors of immunity: ESID registry 1994-2024 report. medRxiv. 2025.
- Vaseghi-Shanjani M, et al. Primary Atopic Disorders: Inborn Errors of Immunity Causing Severe Allergic Disease. Curr Opin Immunol. 2025.
- Castagnoli R, et al. Inborn errors of immunity with atopic phenotypes: A practical guide for allergists. World Allergy Organ J. 2021.
¿Puedo hacerme el estudio de alergia a fármacos si estoy dando lactancia materna?
El estudio de alergia a fármacos es un procedimiento complejo, que incluye una historia clínica alergológica detallada y la realización de pruebas alergológicas como las pruebas in vivo, aquellas que se realizan en el paciente, y las pruebas de laboratorio.
Las pruebas de laboratorio requieren de una muestra de sangre, por lo que no representan un riesgo para los pacientes ni en este caso para mujeres en periodo de lactancia.
Las pruebas in vivo incluyen la realización de tests cutáneos y de la prueba de exposición, que consiste en la administración controlada del fármaco sospechoso. Sin embargo, aunque estas pruebas se realizan según los estándares de seguridad y por personal con experiencia, no están exentas de riesgos y existe la posibilidad de que la paciente presente una reacción alérgica que precise de tratamiento para su resolución. En este sentido, la mayoría de los antihistamínicos modernos (como loratadina, desloratadina o fexofenadina) son seguros durante la lactancia, ya que pasan a la leche en cantidades mínimas. Los corticoides, en dosis bajas, también son compatibles. La adrenalina, aunque puede pasar a la leche y no se conocen del todo sus efectos en el bebé, es un medicamento vital en caso de anafilaxia, por lo que su uso está plenamente justificado. Si se necesita administrarla, lo que se recomienda es suspender temporalmente la lactancia.
Dado que el diagnóstico de confirmación de alergia en ocasiones requiere de su administración, no debería realizarse si el fármaco a estudiar tiene la contraindicación durante la lactancia materna.
En conclusión, sí es posible realizar un estudio de alergia a medicamentos durante la lactancia. Lo fundamental es que el procedimiento lo valore y supervise su alergólogo, y que se tomen las precauciones necesarias para garantizar tanto su seguridad como la del bebé.
Esther Barrionuevo
Especialista en Alergología
Hospital Regional Universitario de Málaga
Alerta seguridad colirio Bilaxten
Alerta de seguridad de la AEMPS: BILAXTEN 6 MG/ML COLIRIO EN SOLUCION, 1 frasco de 5 ml (NR: 88089, CN: 758305).
Retirada del mercado de todas las unidades distribuidas de los lotes afectados y devolución al laboratorio por los cauces habituales.
Certamen Alergia y Humanidades 2025
¿Eres socio de la SEAIC y te gusta escribir o pintar?
Participa en el X Certamen de Poesía, Relato y Pintura y gana un premio de 400 € en cualquiera de sus tres modalidades:
- Poesía
- Relato
- Pintura
Las obras deben estar relacionadas con la alergia.
- Plazo de inscripción: hasta el 15 de septiembre de 2025 a las 14:00 h.
- Formulario de inscripción (debes tener la obra lista para poder subirla en formato digital).
- Las obras físicas se deberán entregar el día 1 de octubre y estarán en exhibición durante el Congreso Nacional SEAIC 2025, que se celebrará del 1 al 4 de octubre en Granada.
- El premio se anunciará en el acto de clausura el 4 de octubre de 2025.
![]() Bases Certamen Alergia y Humanidades 2025 (15530 descargas )
Bases Certamen Alergia y Humanidades 2025 (15530 descargas )
BBI marzo 2025
Boletín bibliográfico de Inmunoterapia del mes de marzo de 2025.
Selección del mes de marzo de 2025, por parte de los miembros del Comité de Inmunoterapia de la SEAIC, de los artículos de inmunoterapia más novedosos y relevantes para la práctica clínica.
Patrocinado por Allergy Therapeutics.
![]() Enlace visible solo para socios de la SEAIC.
Enlace visible solo para socios de la SEAIC.
Si no lo ha hecho, identifíquese aquí.
Congreso Bienal de la Sociedad Internacional de Eosinófilos 2025
13º Congreso Bienal de la Sociedad Internacional de Eosinófilos.
13-16 de Julio – Montpellier (Francia).
El Congreso IES es el foro internacional líder para la investigación de eosinófilos que reúne a expertos del ámbito académico, la práctica clínica y la industria.
Ya está abierto el plazo para registrarse y enviar comunicaciones.
BBI Julio 2024
Boletín bibliográfico de Inmunoterapia del mes de julio 2024.
Selección del mes de julio 2024, por parte de los miembros del Comité de Inmunoterapia de la SEAIC, de los artículos de inmunoterapia más novedosos y relevantes para la práctica clínica.
Patrocinado por Allergy Therapeutics.
![]() Descargas para socios de la SEAIC.
Descargas para socios de la SEAIC.
Si no lo ha hecho, identifíquese aquí.
XX Reunión Anual SESOC
La Sociedad Española de Superficie Ocular y Córnea celebrará su XX reunión anual dedicada a la Alergia Ocular en Madrid, los días 23 y 24 de marzo de 2023. Para más información sobre el programa científico y la inscripción, puede visitar este enlace.
EAACI-ESCD Skin Allergy Meeting 2023
La próxima edición del Skin Allergy Meeting, organizado por la Academia Europea de Alergología e Inmunología Clínica junto a la Sociedad Europea de Dermatitis de Contacto, se celebrará en formato on-line los días 24 y 25 de marzo de 2023. Para más información sobre el programa científico y la inscripción, puede visitar este enlace.